Присоединяйтесь к широкому международному сообществу креативных людей, пользующихся Cliparto каждый день. чтобы покупать или продавать изображения.
| ◢ Мой Cliparto
› ЛайтБокс (0)
Не помните пароль / логин? ◢ Впервые у нас? Зарегистрируйтесь ◢
Есть аккаунт на Vector-Images.
› Недавно просмотрено
| ||||||||||||||||
 Изображения предоставлятся по Royalty-Free лицензиям. Наши Условия использования сервисов разрешают использование изображений для широкого спектра услуг, товаров и отраслей, где изображения, приобретенные через Cliparto будут работать на Вас.
Изображения предоставлятся по Royalty-Free лицензиям. Наши Условия использования сервисов разрешают использование изображений для широкого спектра услуг, товаров и отраслей, где изображения, приобретенные через Cliparto будут работать на Вас.
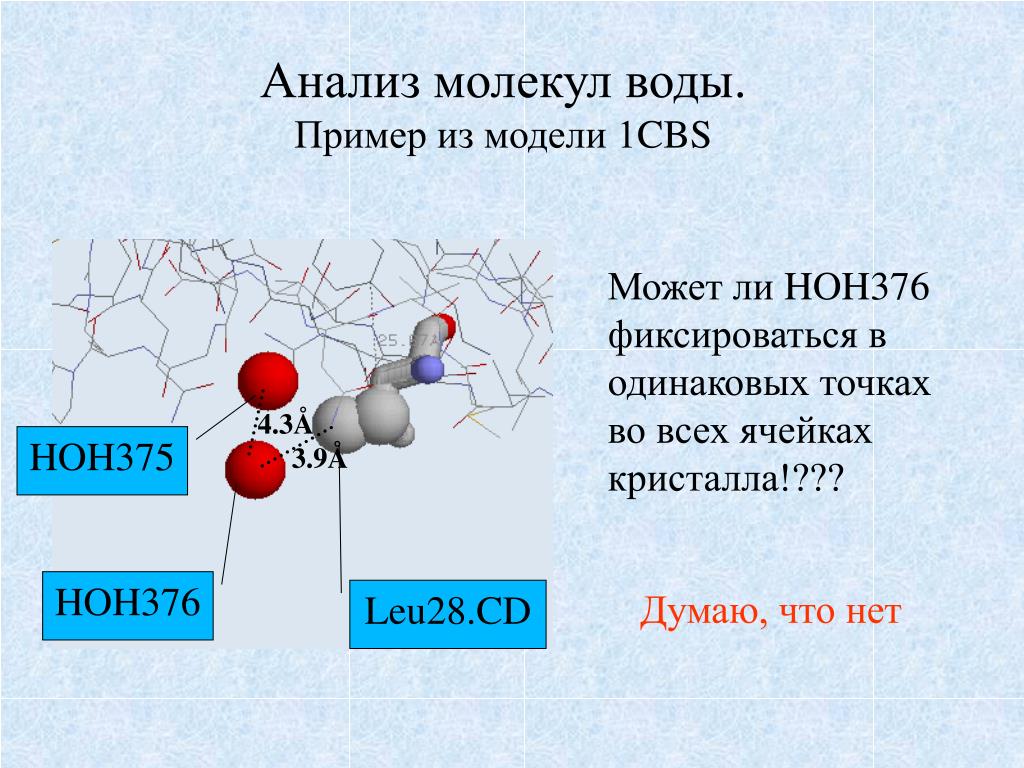
 com?
com?Обои для рабочего стола Модель молекулы на каменной поверхносте фото
НАВИГАЦИЯ: ОБОИ ДЛЯ РАБОЧЕГО СТОЛА >> ОБОИ 3D графика >> Геометрические предметы >> Обои Модель молекулы на каменной поверхносте
Картинку добавил(а): Ulysess Разрешение: 1280 x 1024 Раздел обоев: Скачать похожие обои на Модель молекулы на каменной поверхносте Порекомендовать картинку другу: Ваше имя: |
Похожие обои на Модель молекулы на каменной поверхносте:
Красные молекулыАбстракции
Молекулярная абстракцияАбстракции
Оранжевая молекулаАбстракции
Жёлтая молекула3D графика
Разбитая на молекулы зелёная жидкостьАбстракции
Молекулярные связиАбстракции
Соединения молекул несутся вдоль хромированной дуги3D графика
Многополярная «решётка» из маленьких, круглых молекул и атомовАбстракции
Стека однородных атомных комбинаций с красными молекулкамиАбстракции
Разложенная на молекулы, вибрации и бинарный кодАбстракции
На фоне химической структуры носятся прозрачные молекулкиАбстракции
Голубые молекулы воды в вечном движении океана3D графика
Замёрзшие цепочка ДНК и биологические молекулы3D графика
Макет молекулыМедицина
Молекула ДНКМедицина
Слипшиеся молекулы воды в мореАбстракции
Молекула измеренияАбстракции
Молекула ДНК3D графика
Хаотичное движение молекулАбстракции
Молекула в пространствеАбстракции
Оранжевые молекулы3D графика
Завораживающие молекулы вещества3D графика
Собрания зеленых молекулАбстракции
Органические молекулыРисованные
ХОЧУ ЕЩЕ ТАКИХ ЖЕ ОБОЕВ! >>
Мнения и комментарии к данной картинке
Совет по выбору обоев — «Функциональность» :
Рабочий стол – это то место на компьютере, с которого начинается работа с ним. Не стоит забывать о том, что на рабочем столе, как правило, располагается масса всяческих ярлыков и папок. При выборе обоев для рабочего стола, обратите внимание на то, чтобы на, выбранном вами фоне, не «терялись» полезные ярлыки и ссылки. Ведь какая бы красивая картинка не была, на рабочем столе она может оказаться главным раздражителем, из-за того, что вы не можете найти на экране ссылку на нужную программу или папку. Для того чтобы этого избежать, не следует выбирать для рабочего стола аляповатые, пестрые или же насыщенные деталями изображения, постарайтесь выбрать более «ровную» картинку.
НОВЫЕ ОБОИ НА САЙТЕ
На капотe девушкаДевушки с автомобилями
Спят, кому как нравитсяСобаки и кошки
Босая на ступенькаx сидитДевушки
Сок гранатовый и фрyктыЕда и напитки
Синяя рыбкaЖизнь под водой
Сушится красный перeцЕда и напитки
Во дворикe девочка с собакамиДевушки
Со спины девушка с цветочками синими на волосаxДевушки
NEW! БАНК ОБОЕВ. МИКС
МИКС
Представляем Вашему вниманию Банк Обоев.Микс — удобная возможность выбрать понравившуюся картинку из списка,
который составляется случайным образом!
[lampps-users] трудности с использованием команды molecule для добавления молекулы — Зеркало списка рассылки LAMMPS
Привет,
У меня большие трудности с добавлением молекулы в уже существующую симуляцию. Я старался изо всех сил читать документацию и следовать примерам. Я также делал это раньше с более простой молекулой (газообразный водород), поэтому я озадачен текущими проблемами.
Я использую версию LAMMPS от 7 августа 2019 года; Я хотел бы сохранить эту версию, поскольку я использовал ее для предыдущих симуляций, но при необходимости я могу изменить ее. Я получаю ту же ошибку из версии от 3 марта 2020 года.
Я пытаюсь добавить молекулу, например, в коробку с водой. Сначала я запускаю короткую симуляцию на ящике с водой без проблем. Затем я использую шаблон молекулы, чтобы добавить 1 молекулу наугад в коробку. Я получаю предупреждение и ошибку:
Я получаю предупреждение и ошибку:
ПРЕДУПРЕЖДЕНИЕ: Атрибуты молекулы не соответствуют системным атрибутам (…/molecule.cpp:1386)
ОШИБКА: Топология/атом молекулы превышает системную топологию/атом (…/molecule.cpp:1415)
Я указал необходимое количество дополнительных типов, связей, углов и т.д. Пробовал делать это разными способами. У меня не получается ни в коем случае. Ниже я прикрепил свой шаблон молекулы и сценарий ввода.
Thanks,
Amalie
Here’s the molecule file:
9 atoms
8 bonds
10 angles
Coords
1 -1.603907 1.211837 -0.272400
2 -2.219043 0.545670 0.077819
3 -0.015670 0.068918 -0.020661
4 -0.707721 -1.625115 0.801466
5 -0.391026 -2.153992 0.049244
6 0.932809 -0.511747 -1.667599
7 1.807081 -0.231367 -1.348724
8 1.412678 0.761188 1.181746
9 1.259616 0.068276 1.846429
Types
1 3
2 4
3 5
4 2
5 3
6 2
7 3
8 2
9 3
Charges
1 -1. 0027
0027
2 0.2838
3 0.8756
4 -1.0027
5 0.2838
6 -1.0027
7 0.2838
8 -1.0027
9 0.2838
Masses
1 15.9994
2 1.0078
3 65.4
4 15.9994
5 1.0078
6 15.9994
7 1.0078
8 15.9994
9 1.0078
Bonds
1 1 1 2
2 2 1 3
3 2 3 4
4 2 3 6
5 2 3 8
6 1 4 5
7 1 6 7
8 1 8
У углы
1 1 2 1 3
2 1 5 4
3 1 7 3 6
4 1 9 3 8
5 2 1 3 4
6 2 1 3 6
7 2 1 3 8
8 2 4 3 6
9 2 4 3 8
10 2 6 3 8
Вот входной файл для ламп; файл данных — это просто коробка с водой.
Переменный индекс проекта «Ват»
Единицы Реал
Atom_style Full
PARI_STYLE LJ/CUL/COUL/LONG 10,0 10,0
BOND_STYLE HARMONG
COUL_STYLE HARMONIC
DIHEDRAL_STYLE HARMONG
. нет
нет
read_data вода.данные экстра/атом/типы 3 экстра/связь/типы 2 экстра/угол/типы 2 экстра/специальные/на/атом 20
pair_modify tail №
Kspace_Style PPPM 1.E-4
МАСС 3 15,999
Масса 4 1.0078
Масса 5 65,4
PARE_COEFF 1 1 0.1554 3.16557 # wow, ow
PARIF 1 1 0,1554 3.16557 # wow, ow
Pair_ff_ff_ff_ff_ff 1 0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0.0. HW
PAIR_COEFF 2 2 0,0 0,9572 # HW, HW
PARI_COEFF 3 3 0,2104 3,0664 # AMBER
PARIE_COEFF 4 4 0,0 0,0
BOND_COEFF 5 5 0,0007549,0989 # AMBER
BOND_COEFF 5 5 0,0007599,0989 # AMBER
BOND_COEFF 5 5 0,0007549 2,0989 # AMBER
. 2 700 1,0
bond_coeff 3 700 1.96
angle_coeff 1 75.00000 109.50000 # hw,ow4,hw
angle_coeff 2 1000 94.19
angle_coeff 3 1000 109.47
variable TK equal 300.0
variable PBAR equal 1. 0
0
group wat type 1 2
сосед 1.0 бин
neigh_modify задержка 0 каждый 1 проверка да
скорость все создать 300 3189434 расстояние гауссовское гниение да мама да сумма да
timestep 1.00003
thermo 500
thermo_modify flush yes
fix watsh wat shake 0.0001 20 0 b 1 a 1
fix TPSTAT all npt temp {TK} {TK} 100 iso {PBAR} all custom dump2 1003 900 20000 Вт. LammpStrj ID Mol Type Element x Y Z IX IY IZ
Перезагрузку 1000000 Вт. 0 случайный 1 225698 кл моль цинката 13546
run 10000
write_restart restart.*.lmp
write_data data.*.lmp
molecule command — документация LAMMPS
\(\renewcommand{\AA}}{\text{Å} )
Синтаксис
ID молекулы значения ключевого слова file1 ... значения ключевого слова file2 ... fileN ...
ID = присвоенное пользователем имя для шаблона молекулы
файл1,файл2,… = имена файлов, содержащих описания молекул
Ноль или более пар ключ/значение могут быть добавлены после каждого файла
ключевое слово = смещение или toff или boff или aoff или off или ioff или масштаб
смещение значения = Toff Boff Aoff Doff Ioff Toff = смещение для добавления к типам атомов Boff = смещение для добавления к типам облигаций Aoff = смещение для добавления к типам углов Doff = смещение для добавления к двугранным типам Ioff = смещение для добавления к неправильным типам toff Значение = Toff Toff = смещение для добавления к типам атомов boff значение = Boff Boff = смещение для добавления к типам облигаций aвыкл.
 значение = Aвыкл.
Aoff = смещение для добавления к типам углов
doff Значение = Doff
Doff = смещение для добавления к двугранным типам
iвыкл. значение = Iвыкл.
Ioff = смещение для добавления к неправильным типам
масштаб значение = коэффициент
sfactor = масштабный коэффициент, применяемый к размеру и массе молекулы
значение = Aвыкл.
Aoff = смещение для добавления к типам углов
doff Значение = Doff
Doff = смещение для добавления к двугранным типам
iвыкл. значение = Iвыкл.
Ioff = смещение для добавления к неправильным типам
масштаб значение = коэффициент
sfactor = масштабный коэффициент, применяемый к размеру и массе молекулы
 значение = Aвыкл.
Aoff = смещение для добавления к типам углов
doff Значение = Doff
Doff = смещение для добавления к двугранным типам
iвыкл. значение = Iвыкл.
Ioff = смещение для добавления к неправильным типам
масштаб значение = коэффициент
sfactor = масштабный коэффициент, применяемый к размеру и массе молекулы
значение = Aвыкл.
Aoff = смещение для добавления к типам углов
doff Значение = Doff
Doff = смещение для добавления к двугранным типам
iвыкл. значение = Iвыкл.
Ioff = смещение для добавления к неправильным типам
масштаб значение = коэффициент
sfactor = масштабный коэффициент, применяемый к размеру и массе молекулы Примеры
молекула 1 mymol.txt молекула 1 co2.txt h3o.txt молекула CO2 co2.txt boff 3 aoff 2 молекула 1 mymol.txt смещение 6 9 18 23 14 молекулы объекты файл.1 масштаб 1.5 файл.1 масштаб 2.0 файл.2 масштаб 1.3
Описание
Определите шаблон молекулы, который можно использовать как часть других LAMMPS.
команды, как правило, для определения набора частиц как связанного
молекула или твердое тело. Команды, которые в настоящее время используют молекулу
шаблоны включают:
фиксированный депозит
залить
фиксированный жесткий/маленький
исправить встряхивание
исправить gcmc
зафиксировать/реагировать
create_atoms
Шаблон atom_style
Идентификатор шаблона молекулы может содержать только буквенно-цифровые символы
и подчеркивает.
Один шаблон может содержать несколько молекул, перечисленных по одной в каждом файле.
Некоторые из перечисленных выше команд в настоящее время используют только первый
молекула в шаблоне, и выдаст предупреждение, если шаблон
содержит несколько молекул. Команда шаблона atom_style позволяет шаблонам с несколькими молекулами определять
система с более чем одной шаблонной молекулой.
За каждым именем файла могут следовать необязательные ключевые слова, которые применяются
только к молекуле в файле, используемом в этом шаблоне. Это для
упростить использование одного и того же файла молекулы в другой молекуле
шаблоны или в различных симуляциях. Вы можете указать тот же файл
несколько раз с разными необязательными ключевыми словами.
offset , toff , boff , aoff , doff , ioff ключевые слова
добавить указанные значения смещения к типам атомов, типам связи, углу
типы, двугранные типы и/или неправильные типы, поскольку они читаются из
файл молекулы. Например. если toff = 2, и файл использует типы атомов
Например. если toff = 2, и файл использует типы атомов
1,2,3, то каждая созданная молекула будет иметь типы атомов 3,4,5. Для
ключевое слово offset необходимо указать все пять значений смещения, но
отдельные значения будут игнорироваться, если шаблон молекулы не
использовать этот атрибут (например, без облигаций).
Примечание
Смещения игнорируются в строках, использующих метки типа, как тип
этикетки будут определять фактические типы напрямую в зависимости от
текущие настройки карты меток.
Ключевое слово scale масштабирует размер молекулы. Это может быть
полезен для моделирования полидисперсных гранулированных твердых тел. Масштаб
фактор применяется к каждому из этих свойств в файле молекулы, если
они определены: координаты отдельных частиц (Coords
раздел), индивидуальная масса каждой частицы (раздел Masses),
индивидуальные диаметры каждой частицы (раздел Diameters), общий
масса молекулы (ключевое слово заголовка = масса), центр масс
молекула (заголовок ключевое слово = com), и моменты инерции
молекула (ключевое слово заголовка = инерция).
Примечание
Команда molecule может использоваться для определения молекул со связями,
углы, двугранники, несобственные углы или специальные списки связей соседей
внутри молекулярной топологии, так что вы можете позже добавить молекулы
к вашей симуляции с помощью одной или нескольких команд, перечисленных выше.
Поскольку эта информация, связанная с топологией, требует подходящего хранилища
зарезервирован, когда LAMMPS создает окно имитации (например, при использовании
команда create_box или
команда read_data) необходимо зарезервировать подходящее место
чтобы вы не переполняли эти предварительно выделенные структуры данных при добавлении
молекулы позже. Как команда create_box, так и
команда read_data имеет «дополнительные» параметры, которые
Убедитесь, что пространство выделено для хранения информации о топологии для молекул, которые
добавляются позже.
Формат отдельного файла молекулы аналогичен, но
(не идентичен) файлу данных, прочитанному функцией read_data
команд и заключается в следующем.
Файл молекулы имеет заголовок и тело. Заголовок появляется первым.
первая строка заголовка и, следовательно, файла молекулы , всегда пропускается;
обычно содержит описание файла или комментарий от программного обеспечения
который создал файл.
Затем строки считываются по одной строке за раз. Строки могут иметь окончание
комментарий, начинающийся с «#», который игнорируется. Там должен быть хотя бы один
пробел между допустимым содержимым и комментарием. Если строка пуста
(т.е. содержит только пробел после удаления комментариев), это
пропущено. Если строка содержит ключевое слово заголовка, соответствующий
значение(я) читается/читается из строки. Строка , а не пустая и не
, а не содержит ключевое слово заголовка, с которого начинается тело файла.
Тело файла содержит ноль или более разделов. Первая линия
раздела имеет только ключевое слово. Следующая строка пропускается.
остальные строки раздела содержат значения. Количество строк
зависит от ключевого слова раздела, как описано ниже. Ноль или более пустых
Ноль или более пустых
линии могут быть использованы между разделами. Разделы могут появляться в любом порядке,
за некоторыми исключениями, как указано ниже.
Это распознанные ключевые слова заголовка. Строки заголовка могут входить
Любой заказ. Числовые значения считываются с начала
линия. Ключевое слово должно стоять в конце строки. Все эти
настройки имеют значения по умолчанию, как описано ниже. Линия нужна только
появляются, если значения отличаются от значений по умолчанию.
N атомов = количество атомов N в молекуле, по умолчанию = 0
Nb связей = количество связей Nb в молекуле, по умолчанию = 0
Na углов = количество углов Na в молекуле, по умолчанию = 0
Nd диэдров = количество диэдров Nd в молекуле, по умолчанию = 0
Ni неправильные = количество несобственных Ni в молекуле, по умолчанию = 0
Фрагменты Nf = количество фрагментов в молекуле, по умолчанию = 0
Mtotal масса = общая масса молекулы
Xc Yc Zc com = координаты центра масс молекулы
Ixx Iyy Izz Ixy Ixz Iyz инерция = 6 компонент тензора инерции молекулы
Для масса , ком и инерция , по умолчанию для LAMMPS используется
рассчитать эту величину самостоятельно, если это необходимо, предполагая, что молекулы
состоит из набора точечных частиц или частиц конечного размера (с
ненулевой диаметр), которые не перекрываются. Если частицы конечного размера в
Если частицы конечного размера в
молекулы перекрываются, LAMMPS не будет учитывать перекрытие
эффекты при расчете любой из этих 3 величин, поэтому вы должны
предварительно вычислить их самостоятельно и перечислить значения в файле.
Масса и координаты центра масс (Xc,Yc,Zc)
самоочевидно. 6 моментов инерции (ixx,iyy,izz,ixy,ixz,iyz)
должны быть значения, соответствующие текущей ориентации
твердое тело вокруг своего центра масс. Значения относятся к
оси моделирования XYZ, а не по отношению к главным осям
само твердое тело. LAMMPS выполняет последний расчет
внутри.
Это разрешенные ключевые слова раздела для тела файла.
Координаты, Типы, Молекулы, Фрагменты, Заряды, Диаметры, Массы = разделы свойств атома
Связи, углы, двугранники, несобственности = разделы молекулярной топологии
Подсчет специальных облигаций, специальные облигации = специальная информация о соседе
Флажки встряхивания, атомы встряхивания, типы связи встряхивания = информация о встряхивании
Для разделов «Типы», «Облигации», «Углы», «Двугранники» и «Несобственные
тип атома/связи/угла/и т. д. может быть указан как число (числовой
д. может быть указан как число (числовой
тип) или как буквенно-цифровая метка типа. Последнее допускается только в том случае, если
метки типов были определены либо с помощью команды labelmap, либо в файлах данных, считанных с помощью команды read_data, которые имеют разделы для меток типа Atom, Bond
Типовые метки, метки угловых типов и т. д. См. страницу документации с метками типов, чтобы узнать о разрешенном синтаксисе меток типов.
и общее обсуждение того, как можно использовать метки типов.
При использовании меток типа любые значения, указанные как со смещением игнорируются.
Если указан раздел «Облигации», то количество специальных облигаций и
Разделы «Специальные облигации» также можно использовать, если это необходимо, для явного
перечислите 1-2, 1-3, 1-4 соседей в топологии молекулы (см.
подробности ниже). Это необязательно, так как если эти разделы не
включены, LAMMPS автоматически сгенерирует эту информацию. Обратите внимание, что
LAMMPS использует эту информацию для правильного исключения или взвешивания попарно связанных
взаимодействия между связанными атомами. Подробнее см. команду special_bonds. Одна из причин перечислить
Подробнее см. команду special_bonds. Одна из причин перечислить
специальная информация об облигациях явно предназначена для термализованного друда
модель осциллятора, которая рассматривает связи между ядерными
ядра и друдовские электроны по-разному.
Примечание
Требуется ли раздел, зависит от того, как шаблон молекулы
используется другими командами LAMMPS. Например, чтобы добавить молекулу
через команду фиксированного депозита, координаты и
Разделы типов обязательны. Чтобы добавить твердое тело через исправление
команды заливки разделы «Связи» (углы и т. д.) не
требуется, так как молекула будет рассматриваться как твердое тело. Немного
разделы необязательны. Например, заливка фикса
Команда может использоваться для добавления «молекул», которые представляют собой кластеры
гранулированные частицы конечного размера. Если раздел Диаметры не
указано, каждая частица в молекуле будет иметь значение по умолчанию
диаметр 1,0. См. страницы документации для команд LAMMPS, которые используют
шаблоны молекул для более подробной информации.
Все разделы перечислены ниже в алфавитном порядке. Формат каждого
описывается раздел, включая количество строк, которые он должен содержать, и
правила (если есть) того, может ли он появиться в файле данных. Для пер-
разделы атомов, записи должны быть пронумерованы от 1 до атомов (где
Natoms — количество атомов в шаблоне), указывающее, какой атом
(или облигации и т. д.), к которым относится запись. Поатомные секции должны
включать настройку для каждого атома, но атомы могут быть перечислены в любом
заказ.
Координаты секция:
одна строка на атом
синтаксис строки: ID x y z
x,y,z = координата атома
Тип Раздел:
Молекулы Раздел:
Фрагменты Раздел:
одна строка на фрагмент
синтаксис строки: ID a b c d …
a,b,c,d,… = идентификаторы атомов во фрагменте
Идентификатор фрагмента может содержать только буквенно-цифровые символы и
подчеркивает. Идентификаторы атомов должны быть значениями от 1 до атомов, где
Идентификаторы атомов должны быть значениями от 1 до атомов, где
Natoms = количество атомов в молекуле.
Сборы Раздел:
одна строка на атом
синтаксис строки: ID q
q = заряд атома
Этот раздел разрешен только для стилей атомов,
плата за поддержку. Если этот раздел не включен, плата по умолчанию
на каждый атом в молекуле 0,0.
Диаметры сечение:
Этот раздел разрешен только для стилей атомов,
поддерживают сферические частицы конечного размера, т.е. сфера atom_style. Если
не указан, диаметр каждого атома в молекуле по умолчанию равен 1,0.
Массы Секция:
Этот раздел разрешен только для стилей атомов,
поддерживают массу на атом, а не массу на тип. См.
массовая команда для деталей. Если этот раздел не
включая, масса по умолчанию для каждого атома получается из его объема
(см. раздел «Диаметры») и плотностью по умолчанию 1,0 в
единицы массы/объема.
Облигации раздел:
одна строка на облигацию
синтаксис строки: тип идентификатора atom1 atom2
type = тип связки (1-Nbondtype или этикетка с указанием типа)
атом1, атом2 = ID атомов в связи
Идентификаторы двух атомов в каждой связи должны быть значениями
от 1 до Natoms, где Natoms = количество атомов в молекуле.
Уголки Секция:
одна линия на угол
синтаксис строки: тип ID атом1 атом2 атом3
тип = угловой тип (1-Nangletype или типовая табличка)
atom1,atom2,atom3 = ID атомов в углу
ID для трех атомов в каждом угле должны иметь значения от 1 до
Natoms, где Natoms = количество атомов в молекуле. 3 атома
упорядочены линейно внутри угла. Таким образом, центральный атом (вокруг
который вычисляет угол) является атомом2 в списке.
Диэдры сечение:
одна линия на двугранник
синтаксис строки: тип ID атом1 атом2 атом3 атом4
- Тип
= двугранный тип (1-N двухгранный тип или табличка с указанием типа)
атом1,атом2,атом3,атом4 = идентификаторы атомов в диэдре
ID для четырех атомов в каждом двуграннике должны быть значениями от 1 до
Natoms, где Natoms = количество атомов в молекуле. 4 атома
4 атома
упорядочены линейно внутри диэдра.
Неправильные элементы Раздел:
одна строка на неправильный
синтаксис строки: тип ID атом1 атом2 атом3 атом4
type = неправильный тип (1-Nimpropertype или типовая этикетка)
atom1,atom2,atom3,atom4 = идентификаторы атомов в неправильных
Идентификаторы четырех атомов в каждом несобственном элементе должны иметь значения от 1 до
Natoms, где Natoms = количество атомов в молекуле. Заказ
4 атома определяют определение неправильного угла, используемого в
формула для определенного неправильного стиля. Видеть
страницы документа для отдельных стилей для деталей.
Специальные счета для облигаций Раздел:
N1, N2, N3 — количество 1-2, 1-3, 1-4 соседей соответственно
этот атом в топологии молекулы. См.
страница special_bonds для более подробного обсуждения
1-2, 1-3, 1-4 соседи. Если появится этот раздел, специальные облигации
также должен появиться раздел.
Как объяснялось выше, LAMMPS автоматически сгенерирует эту информацию, если
раздел не указан. Если указано, этот раздел будет
переопределить то, что будет сгенерировано автоматически.
Специальные облигации Раздел:
одна строка на атом
синтаксис строки: ID a b c d …
a,b,c,d,… = идентификаторы атомов в специальных связях N1+N2+N3
A, b, c, d и т. д. — идентификаторы атомов n1+n2+n3, равные 1–2, 1–3,
1-4 соседа этого атома. Идентификаторы должны быть значениями от 1 до
Natoms, где Natoms = количество атомов в молекуле. Первый N1
значения должны быть 1-2 соседями, следующий N2 должен быть 1-3
соседи, последние N3 должны быть 1-4 соседями. Идентификатор атома не должен
появляются более одного раза. См. документ special_bonds
страница для более подробного обсуждения 1-2, 1-3, 1-4 соседей. Если этот раздел
появится раздел Special Bond Counts.
Как объяснялось выше, LAMMPS автоматически сгенерирует эту информацию, если
раздел не указан. Если указано, этот раздел будет переопределять
Если указано, этот раздел будет переопределять
что будет автоматически сгенерировано.
Встряхивающие флажки Секция:
Этот раздел необходим только в том случае, если молекулы созданы с использованием шаблона
будет ограничиваться SHAKE с помощью команды «fix shake». Другой
два раздела Shake также должны появиться в файле после этого.
Значение флага для каждого атома следующее. См. страницу исправления встряхивания для дальнейшего описания SHAKE.
кластеры.
0 = не является частью кластера SHAKE
1 = часть углового кластера SHAKE (две связи и угол, который они образуют)
2 = часть 2-атомного кластера SHAKE с одинарной связью
3 = часть трехатомного кластера SHAKE с двумя связями
4 = часть 4-атомного кластера SHAKE с тремя связями
Shake Atoms Раздел:
одна строка на атом
синтаксис строки: ID a b c d
a,b,c,d = идентификаторы атомов в кластере
Этот раздел необходим только в том случае, если молекулы созданы с использованием шаблона
будет ограничиваться SHAKE с помощью команды «fix shake». Другой
Другой
в файле также должны присутствовать два раздела Shake.
Значения a,b,c,d представляют собой идентификаторы атомов (от 1 до Natoms) для всех атомов
в кластере SHAKE, к которому принадлежит этот атом. Количество значений
которое должно появиться, определяется флагом встряхивания атома (см.
раздел «Встряхнуть флаги» выше). Все атомы в конкретном кластере должны
перечислите их значения a,b,c,d одинаково.
Если флаг = 0, в строке не указаны значения a,b,c,d, только
(игнорируется) ИД.
Если флаг = 1, перечисляются a,b,c, где a = ID центрального атома в
угол, а b, c два других атома в углу.
Если флаг = 2, перечисляются a,b, где a = идентификатор атома, связанного с
самый низкий ID, а b = ID атома, связанного с самым высоким ID.
Если флаг = 3, перечисляются a,b,c, где a = ID центрального атома,
b,c = ID двух других атомов, связанных с центральным атомом.
Если флаг = 4, перечисляются a,b,c,d, где a = ID центрального атома,
и b, c, d = ID трех других атомов, связанных с центральным атомом.
Подробное описание см. на странице исправления встряхивания.
кластеров SHAKE.
Shake Bond Types Секция:
одна строка на атом
синтаксис строки: ID a b c
a,b,c = типы связей (или тип угла) связей (или угла) в кластере
Этот раздел необходим только в том случае, если молекулы созданы с использованием шаблона
будет ограничиваться SHAKE с помощью команды «fix shake». Другой
в файле также должны присутствовать два раздела Shake.
Значения a,b,c представляют собой типы облигаций для всех облигаций в кластере SHAKE, которые
принадлежит этот атом. Типы облигаций могут быть любыми числами (от 1 до Nbondtypes)
или метки типа связи, как определено командой labelmap
или раздел «Ярлыки типов облигаций» файла данных.
Количество значений, которые должны появиться, определяется флагом встряхивания
для атома (см. выше раздел Shake Flags). Все атомы в
конкретный кластер должен перечислять свои значения a, b, c одинаково.
Если флаг = 0, в строке не указаны значения a,b,c, только
(игнорируется) ИД.
Если флаг = 1, перечисляются a,b,c, где a = тип связи между
центральный атом и первый нецентральный атом (значение b в Shake
раздел атомов), b = тип связи между центральным атомом и
второй нецентральный атом (значение c в разделе Shake Atoms) и c
= тип угла (от 1 до Nangletypes или метка типа угла) угла
между 3 атомами.
Если флаг = 2, указывается только a, где a = тип связи между
2 атома в кластере.
Если флаг = 3, перечисляются a,b, где a = тип связи между
центральный атом и первый нецентральный атом (значение b в Shake
раздел атомов), а b = тип связи между центральным атомом
и второй нецентральный атом (значение c в разделе Shake Atoms).
Если флаг = 4, перечисляются a,b,c, где a = тип связи между
центральный атом и первый нецентральный атом (значение b в Shake
раздел атомов), b = тип связи между центральным атомом и
второй нецентральный атом (значение c в разделе Shake Atoms) и c
= тип связи между центральным атомом и третьим
нецентральный атом (значение d в разделе Shake Atoms).